This article will be based on the first three issues of the "problem interpretation series", combined with a very representative article (Nature methods), a comprehensive and in-depth analysis: the quality characterization of gene knockout (fragment loss or shift mutation) cell lines obtained through CRISRP-Cas9, to better assist basic scientific research and drug development. Through the case analysis, further analysis of the answer: why protein spectrum verification is the gold standard for KO cell knockout verification.
WB is a comprehensive immunological detection technology, which uses specific antibodies to perform gel electrophoresis on cells or biological tissue samples. It uses SDS-PAGE technology to separate protein molecules in biological samples on the gel according to their molecular weight, and then transfers the protein to the solid phase membrane by electrotransfer. The protein on the solid phase membrane is used as antigen to react with the corresponding antibody, and enzyme-labeled second antibody reaction, through the substrate color or fluorescence imaging methods to detect electrophoresis separation of specific target gene expression of protein. This method is heavily dependent on the quality, especially the specificity, of the monoclonal antibody in KO cell knockout validation.
The Elem team recommend uses KO-verified monoclonal antibodies (welcome to visit Elem's official website for inquiries) for WB detection of KO cells and WT cells (wild-type cells), which can obtain more reliable verification data. If there is no suitable KO verification antibody, it is recommended to use protein spectrum to verify KO.
2, Protein Spectrum Experiment
Protein spectrum experiment is through the complex proteome samples (such as cell precipitation, tissue, body fluid, etc.) protein extraction, separation, enzyme digestion, peptide separation, mass spectrometry data collection and search library, can be at the same time thousands of proteins in the sample qualitative or quantitative analysis, so as to determine the type and relative abundance information of proteins in different samples. The main principle is that the protein is digested by protease into peptide mixture, and the peptide mixture is ionized to form charged ions in the mass spectrometer. The electric field and magnetic field of the mass spectrometer separate the peptide ions with a specific mass to charge ratio (I. e. mass to charge ratio, m/z). The separated ions are collected by the detector to determine the m/z value of each ion, proteins are identified by software alignment of the primary and secondary mass spectral peaks generated after trypsin digestion with a theoretical database (amino acid sequences downloaded from uniprot or converted from transcriptome data). Mass spectrometry can accurately identify and quantify all proteins expressed in a genome or all proteins in a complex mixed system.
After proteomic pre-processing, data collection and data analysis of KO cell pellets and WT cells/control cells at the same time, the deletion of polypeptides of target genes and the changes of related proteins (volcano map) can be analyzed: the knockout of target genes and the changes of related metabolic pathways caused by the deletion of target genes.
2. classic literature sharing
With the development of CRISPR-Cas9-based technologies, gene editing has become a widely used tool in the field of biological research. The technology relies on Cas9, a nuclease guided by a short-strand guide RNA(sgRNA), which precisely cuts the DNA double strand of the coding region of the target gene under its guidance, thereby achieving genetic knockout (KO). When the Cas9-sgRNA complex (I. e., RNP) acts on a specific sequence, DNA double-strand breaks will be repaired through the NHEJ pathway (non-homologous end joining repair pathway). This process is error-prone and may lead to insertion or deletion mutations, which in turn lead to frameshift mutations and premature termination codons (PTC), which eventually lead to nonsense-mediated degradation (NMD) of mRNA and degradation of abnormal peptide chain products.
Nonsense-mediated RNA degradation (NMD) is a key post-transcriptional monitoring mechanism responsible for recognizing and degrading mRNA containing premature termination codons (PTC). This mechanism aims to prevent the translation of these abnormal mRNAs into potentially toxic truncated proteins, thereby preventing the accumulation of harmful proteins in the cell.
The mechanism of NMD is widespread and highly conserved in eukaryotic cells, ranging from unicellular yeast to complex multicellular organisms such as nematodes, plants and animals, and its wide distribution suggests that NMD plays an indispensable role in maintaining genomic stability and normal gene expression.
In addition to degrading PTC-containing mRNAs produced by mutations, NMD is also involved in the regulation of other types of transcripts, such as mRNAs containing upstream open reading frames (u-ORF), mRNAs with very long 3 'untranslated regions, and even certain non-coding RNAs. Studies have shown that NMD plays a role in the regulation of mRNA expression in about 25% of the body, involving physiological and pathological processes such as cell differentiation, stress response, some genetic diseases and the occurrence and development of tumors.
Many studies have explored CRISPR-Cas9 possible off-target effects, and in order to improve the specificity of the CRISPR-Cas9, researchers have implemented improved strategies including the use of Cas9 nuclease variants, the optimization of sgRNA design, and the protein engineering of the nuclease itself. However, there is a lack of research progress on whether frameshift mutations lead to the expected complete loss of protein expression and activity.
Researchers from the European Molecular Biology Laboratory in Germany, Wolfgang Huber, Lars M. Steinmetz, and GlaxoSmithKline Gerard Drewes, published online in Nature Methods entitled: Biological plasticity can rescue target activity in CRISPR knockout (Biological plasticity rescues target activity in CRISPR knock outs)of the article. The authors combined the experimental strategy of RNA sequencing with tertiary mass spectrometry analysis, combined with quantitative transcriptomics and proteomics studies, to characterize the deletion of 193 targeted 136 different genes generated by CRISPR-induced frameshift in HAP1 cells. Approximately 1/3 of the quantified targets were found to exhibit residual protein expression, with levels ranging from low to primitive, and two causal mechanisms were identified: Translational reinitiation leads to N-terminal truncation of target proteins, or Be Skipping of editing exons leads to protein isoforms with internal sequence deletions. Detailed analysis of the three truncated targets, BRD4, DNMT1 and NGLY1, showed that protein function was partially preserved. Therefore, For CRISPR-Cas9 generated KO cell lines, systematic characterization of residual protein expression or function is necessary to explain the phenotype.
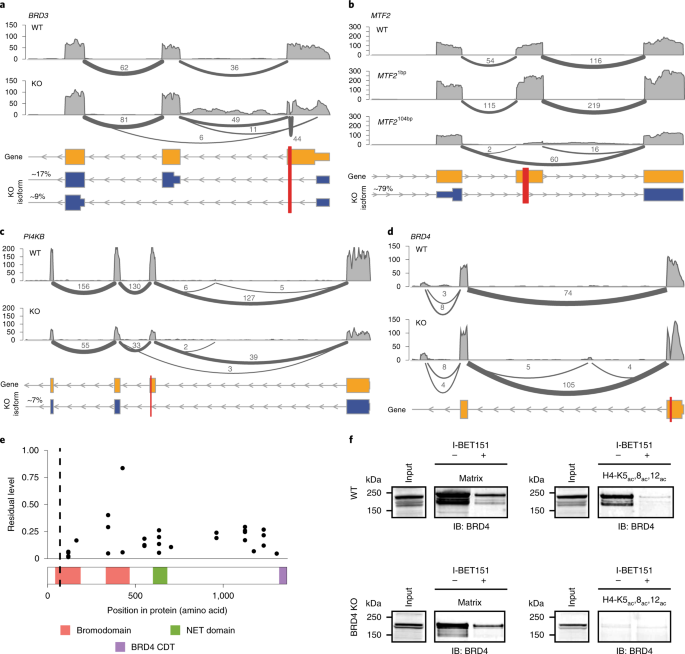
1 Residual transcripts and protein expression in 193 HAP1 cell lines carrying frameshift KO mutations
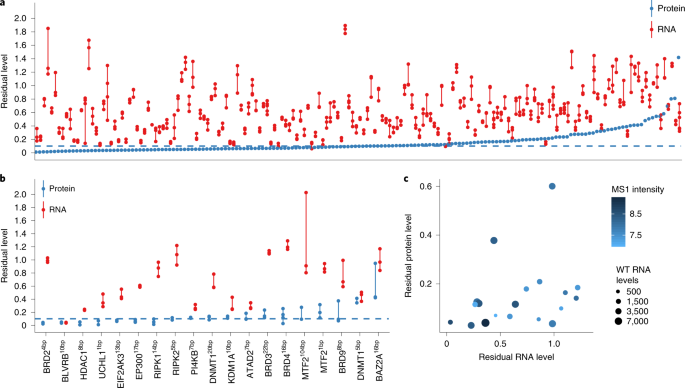
First, the author studied 193 HAP1 knockout cell lines (the HAP-1 knockout cell is derived from a near-haploid human cell line HAP1, which contains a single copy of most genomic sites and is an attractive model system that can be used to track genome editing events. The Elem team will also have a targeted introduction). Each cell line contains 1 gene knockout of 136 genes that are CRISPR-Cas9 targeted to induce frameset KO mutations. In each KO line, the authors calculated residual level expression in the KO and parental lines (wild type) using 3 ′ RNA sequencing (3 ′-RNA-sequence; FIG. 1a) and taking into account transcripts from target genes. A residual level of 0 indicates complete elimination, and a value of 1 indicates that the target transcript level is not different from the parental line.
Contrary to the expectations of the authors, the 174 residual levels are indeed not concentrated around 0, but cover a wide range. Thus, mRNA was not strongly reduced in many KO mutants.The presence of PTC (premature stop codon) was confirmed by DNA sequencing, implying that NMD did not respond or reacted weakly to these transcripts and that the efficiency of NMD depended on complex biological factors.
In-depth analysis of target proteins using tandem mass tag (TMT) isobaric labeling combined with tertiary mass spectrometry (MS3) found that about 1/3 of the KO lines exhibited residual protein levels above this threshold (as shown in Figure 1a), and there was no significant correlation between residual RNA and protein levels, indicating that RNA levels cannot predict protein levels. To further validate this finding, 19 lines were selected for quantitative analysis of transcript RNA-seq and MS3 (as shown in Figure 1b). These quantitative results were verified by at least two repetitions with good repeatability. Consistent with the observations in Figure 1, there was no significant correlation between residual RNA and protein levels. Furthermore, the residual levels did not depend on their initial levels before CRISPR modification, I .e., were not affected by RNA-seq readings and MS1 intensity (determined by the TOP3 method) (as shown in Figure 1c).
1b, KO cell lines of MTF2104bp, PI4KB and BRD3 exhibited residual protein levels of 9% to 28%. Among them, the transcription level of PI4KB decreased, while the transcription level of MTF2104bp and BRD3 was similar to that of wild type.
To explore this phenomenon, the author analyzes Do these mutant lines express alternative transcripts that skip the editing exon.
Figure 2 Exon skipping or translation restart induced residual protein expression
In the BRD3 KO line, there is a splicing event in the BRD3 transcript that spans from the 5 'untranslated region (UTR) to the second or third coding exon (Figure 2a). This splicing results in the deletion of the start codon and may trigger a frameshift mutation. However, our data indicate that by extending the 5 'UTR and using Met81 or Met125 as alternative start codons, N-terminally truncated BRD3 proteins can be generated.
In MTF2104bp cell line, the insertion of 104 base pairs (bp) and the associated frameshift resulted in a significant alternative splicing event (Figure 2b). In particular, exons containing frameshift mutations are often skipped. Notably, this splicing pattern did not occur in the MTF2 mutant line that used the same sgRNA but inserted 1bp (Figure 2b). The PI4KB frameshift mutation resulted in low-level expression of a splice variant that skipped the frameshift-containing exon (Figure 2c), resulting in an in-frame deletion of 228 bp(76 amino acids). This splicing event occurs only in-7% of PI4KB transcripts. This result shows that, Low levels of residual protein from alternatively spliced transcripts.
The BRD4 KO series carried a 16bp deletion in the exon. Although there was residual RNA and protein expression (Figure 1b), no evidence of exon skipping was found (Figure 2d). In order to further characterize the residual protein, quantitative analysis of tryptic peptides was performed on different regions of BRD4, and the results showed that the N-terminal region of the mutant was expressed at a lower level, corresponding to the exon target region before and after CRISPR-Cas9 editing (Figure 2e). However, the residual expression level of the target site region in the C- terminal direction exceeded 10%. The N-terminal truncation of BRD4 may be due to translation restarting at Met105, Met107, or Met132. Notably, Met105 is highly conserved in the bromodomain protein family and there is a putative isoform in BRD2 that is expressed starting from the methionine corresponding to Met105 in BRD4 (Figure 2f). In addition, one of the identified BRD3 truncations also starts at Met81, which corresponds to Met105 in BRD4. This truncated subtype lacks the first 30 amino acids of the first bromodomain (BD1), which is essential for the binding of BRD4 to acetylated histone H4. To test whether the truncated protein retained any function, the group performed a histone H4 binding assay. The results show that, Although the truncated BRD4 can I-BET bind to the small molecule bromodomain inhibitor, it cannot bind to the triacetylated histone H4 tail peptide, which binds to BD1. These results confirm the deletion of BD1 and the retention of functional BD2 in the BRD4 KO.
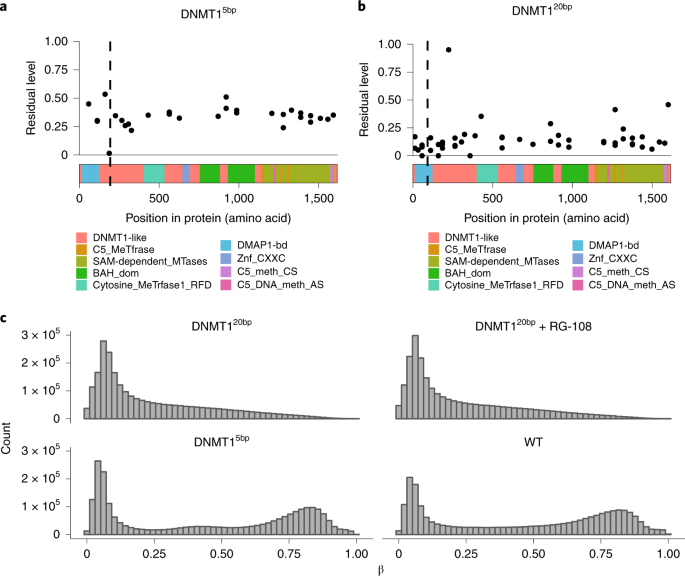
Figure 3 Residual protein expression leads to retention of DNMT1 activity in one of the two KO lines
The expression of these two DNMT1 KO lines was determined by RNA-seq and proteomics (Figure 1b). The DNMT15bp line had a 5bp deletion in the exon, and peptide level analysis showed about 40% residual expression of the entire gene body (Figure 3a). Although three reads supporting skipping were observed to span a 79bp long CRISPR targeting exon, which may have resulted in a frameshift of the spliced transcript, the expression pattern at the peptide level better explains the presence of the two truncated proteins. The deletion directly results in the presence of a stop codon downstream of the CRISPR target site, resulting in a C- terminally truncated protein product consisting of the first 188 amino acids. DNMT120bp lines had a 20bp deletion in exon 4, but no evidence of excision of the targeted exon was found. The residual peptide content is nearly uniform, about 10% of the entire gene body (Figure 3b), which is close to the noise threshold established in the MS3 benchmark.
To assess the residual protein activity of the two mutant lines, genome-wide methylation levels were measured using a methylation array. DNMT1 acts as a major protein that maintains DNA methylation, copying pre-existing methylation markers onto hemimethylated DNA strands during DNA synthesis. The DNMT1 5bp line showed similar methylation levels as the wild type (Figure 3c), consistent with residual protein activity. In contrast, the DNMT120bp line showed little evidence of DNA methylation (Figure 3c). RG-108 treatment of DNMT120bp lines with DNMT1 inhibitors did not alter their methylation levels, consistent with having completely abolished DNMT1 methylation activity. These results indicate that DNMT15bp KO lines have residual protein activity.
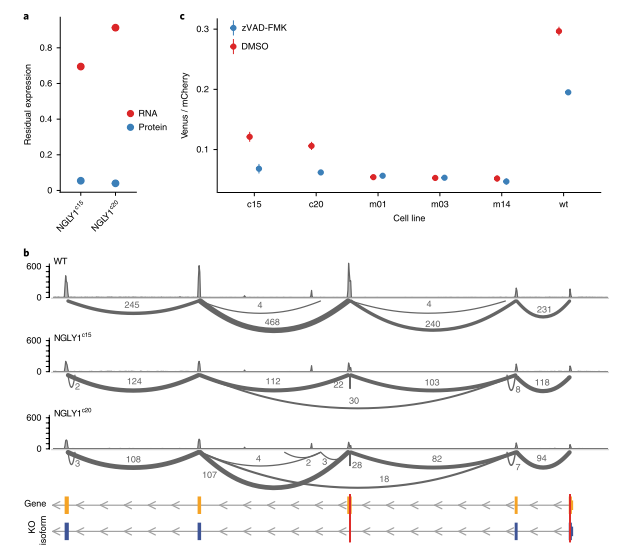
Figure 4 NGLY1 frameshift mutation in K562 cells leads to partial functional NGLY1 truncation
To further expand the analysis to a second cell type, a frameshift KO mutation in the NGLY1 gene, which encodes deN-glycosylase (Figure 4), in K562 cells, successfully isolated two KO clones, designated NGLY1c15 and NGLY1c20, which were derived from the expansion of a single KO cell. Expression of the residual target protein could not be detected by quantitative mass spectrometry (Figure 4a). RNA-seq analysis revealed that exon skipping occurred in both clones and contained frameshift mutations (Figure 4b). Skipping of this exon results in an in-frame deletion of 246bp, such that the truncated protein lacks the amino acid sequence from Gly74 to Thr155.
Analysis of the deglycosylation activity of the target enzyme showed that the NGLY1 KO series showed deglycosylation activity, although its activity was reduced to 60-65% compared to wild-type cells (Figure 4c). Deglycosylation activity was further reduced when NGLY1 inhibitor zVAD-FMK was added (Figure 4c). In order to exclude the non-specific effect of zVAD-FMK, analysis of patient-derived mutant cell lines showed that the NGLY1 exon in these cell lines had a frameshift mutation, which completely eliminated the deglycosylation activity (Figure 4d). Notably, in these clones, zVAD-FMK treatment did not further reduce deglycosylation activity (Figure 4c). These results indicate that NGLY1c15 and NGLY1c20 clones retain some measurable NGLY1-dependent deglycosylation activity despite their reduced cellular deglycosylation activity.
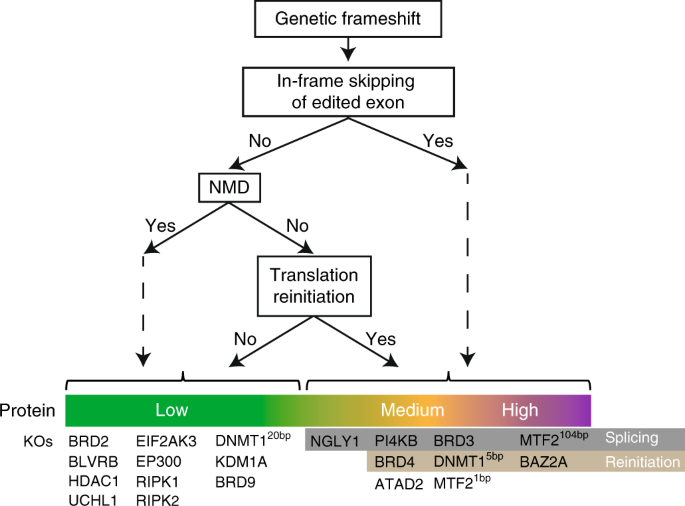
Figure 5 Consequences of CRISPR-Cas9-generated frameshift mutations
The authors summarize the findings in a model that details the possible effects of frameshift KO mutations (Figure 5). The first interesting observation is that Heterogeneity in the efficiency of nonsense-mediated mRNA degradation (NMD) in KO lines. Alternative splicing to exclude premature termination codons (PTC) in the transcript, as well as translational restarts, are all responsible for incomplete NMD efficiency. Notably, even in lines where residual protein expression could not be detected, NMD efficiency (as assessed by residual RNA levels) remained variable. This observation is consistent with a recent study that reported multiple mechanisms affecting NMD efficiency and showed that frameshift mutations can lead to protein production, although NMD effects are weak or non-existent.
This paper expounds the potential limitations of CRISPR-Cas9 technology in creating gene knockout models, especially in the functional consequences after gene editing, suggesting that it is necessary for us to pay attention to and deeply explore the unintended effects and residual protein functions brought by this gene editing tool in practical applications in order to promote the accuracy and safety of gene editing technology.
3. how to efficiently obtain loss-of-function KO cells
1. Description of regular gene knockout strategies
The CRISPR-Cas9 gene knockout system is an efficient gene editing tool that uses Cas9 endonuclease to precisely cut double-stranded DNA. When double-stranded DNA is cleaved by the Cas9 enzyme, the organism initiates the non-homologous end-joining repair pathway (NHEJ) to repair these breaks. In order to achieve precise gene knockout,The regular gene knockout strategy is to carefully design 2-3 guide RNAs (guide RNA1 and guide RNA2) in the upstream and downstream regions of the gene to be knocked out, respectively in multiple exon regions of the target gene.These guide RNAs are introduced into the cell along with a plasmid carrying the gene encoding the Cas9 protein, and the guide RNAs precisely target the target DNA region near the PAM sequence through complementary base pairing. Once the binding is successful, the Cas9 protein cuts the DNA double strand at that location, causing a double-strand break. Organisms have a natural DNA damage repair mechanism, and when a double strand of DNA breaks, a series of repair processes are initiated. In the NHEJ repair pathway, several bases may be inserted or deleted from the DNA at the break, and subsequently, the ends of the broken double-stranded DNA are rejoined together. This series of repair process eventually leads to the target gene is knocked out, so as to achieve the silencing of the gene.
The relevant issues analyzed in the above article are the important risks to be considered in gene knockout strategies based on this regular.
2. Gene knockout strategy for Elem optimization
(1) By targeting the early exons of the encoded functional protein, the Elem realizes the knockout strategy of non -3-fold shift mutation and large fragment loss at the same time, so as to reduce the occurrence probability of N-terminal and C- terminal truncated proteins;
(2) By targeting the shared exons of multiple isoforms of the target gene, the Elem further reduces the probability of the occurrence of "protein subtypes with internal sequence deletions due to the skipping of edited exons;
(3) Elem integrates Sanger sequencing, WB validation and proteome mass spectrometry and other validation methods to comprehensively evaluate genomic low-off-target analysis and provide a high level of validation of gene knockout monoclonal homozygotes.
(4) Based on the unique gene knockout strategy, the Elem team has completed the knockout verification of 19883 genes in the human genome in primary cells, and has continuously increased the investment in research and development at the gene level and protein level verification to provide high-quality verification for customers and the in vitro eukaryotic cell gene knockout subdivision industry ( “ Qizi Verification Quality Standard ”) methods and standards are recognized and practiced by more and more industry experts and scholars!
4. protein profiling becomes the detection of truncated proteins andEffective means of editing exon skipping phenomenon
Why protein profiling is the gold standard for KO cell knockout validation:
1. Wide range of application: mass spectrometry does not require highly specific antibodies, making the method more extensive and easy to apply to the knockout verification of any target protein.
2. There are many types of proteins that can be analyzed: each mass spectrometry experiment can simultaneously analyze a large number of proteins (including target proteins and their interacting proteins, etc.), not only analyzing gene editing target proteins, but also off-target analysis.
3. Accurate quantification: mass spectrometry can be used for quantitative analysis. Although WB results can also reflect the difference of protein in the sample, it can only roughly judge the difference of protein amount. Mass spectrometry method can be more accurate detection, more intuitive to see the protein differences in different samples.
4. Detectable truncation protein: For truncation protein at N-terminal or C- terminal, mass spectrometry can usually cover detection. Compared with the area (8-10 amino acids) covered by epitope of WB experiment, mass spectrometry method is more convenient to see global information. At the same time, it is more realistic than the transcriptome data (the above article proves that there is no obvious correlation between the residual RNA after gene knockout and the protein level).
The application of gene knockout technology is more and more extensive. Whether the gene in the coding region or the non-coding region is knocked out, the final experimental materials will be used for related research and verification. The overall quality characterization of gene knockout cells is as important as the design strategy of gene knockout!
The Elem team hopes to communicate your research background and purpose in depth, provide you with more professional and personalized gene editing programs and verification methods, and help you with basic research and drug development!